
David Welch
Department of Computer Science
Computational evolution group
Room 365 Building 303S
Ph + 64 9 923 8930
david.welch@auckland.ac.nz
My University profile
My Google scholar profile
On Twitter
About me
I am a Senior Lecturer in the Department of Computer Science at the University of Auckland and a principal investigator in the Computational Evolution Group.
My research interests include developing and fitting phylogenetic models to epidemiology (so-called "phylodynamics"), Bayesian computational methods including MCMC and ABC, epidemics on networks, stochastic simulation and inference for coalescent-based population genetics.
I currently teach courses in computational biology and inference, and bioinformatics. Feel free to contact me about research projects offered at Honours, Masters and PhD level.
News
- Jul 2015: Talk on estimating neighbourhood size at MMEE 2015 in Paris (slides) and sampled ancestors at SMBE 2015 in Vienna (slides).
- Dec 2014: Promoted to Senior Lecturer (see Auckland academic payscale here)
- Jan 2014: Co-organised the 18th Annual New Zealand Phylogenomics meeting on the lovely Waiheke Island
- Oct 2013: PI on new Marsden grant, "A unified framework for phylodynamic inference of infectious diseases" with Alexei Drummond and Tanja Stadler
- Jul 2013: PI on new FRDF grant, "Bayesian phylodynamics for infectious diseases" with Alexei Drummond and Tim Vaughan
Academic bio
- Senior Lecturer, Department of Computer Science, University of Auckland, 2015-
- Lecturer, Department of Computer Science, University of Auckland, 2011-2014
- Postdoctoral Researcher, Centre for Disease Dynamics and Department of Statistics, Penn State University, USA, 2008-2011
- Postdoctoral Researcher, Imperial College London, UK, working with David Balding and Mark Beaumont, 2006-2008
- Ph.D, Applied Mathematics, University of Auckland, supervised by Geoff Nicholls, Allen Rodrigo and Wiremu Solomon, 2006
- B.A. (Maori) and B.Sc. Hons. (Maths), University of Otago.
Teaching
- Computational Science (CompSci 369), 2012-2015
- Comparative Bioinformatics (BioInf 702), 2012-2015
- Genome Bioinformatics and Systems Biology (BioInf 703), 2012-2015
- Discrete Structures in Mathematics and Computer Science (CompSci 225), 2013
- Algorithms and Data Structures (CompSci 220), 2012
- Elementary Statistics (Stat 200), Penn State, 2011
Publications
Also see listings at Google scholar
|
Bayesian total evidence dating reveals the recent crown radiation of penguins
arXiv preprint arXiv:1506.04797, 2015
The total evidence approach to divergence-time dating uses molecular and morphological data of extant and fossil species to infer phylogenetic relationships, species divergence times, and macroevolutionary parameters in a single coherent framework. Current model-based implementations of this approach lack an appropriate model for the tree describing the diversification and fossilisation process which can result in erroneous conclusions. We address this shortcoming by providing a total evidence method implemented in a Bayesian framework. This approach uses a mechanistic tree prior to describe the underlying diversification process that generated the tree of extant and fossil taxa. Previous attempts to apply the total-evidence approach have used tree priors that do not account for the possibility that fossil samples may be direct ancestors of other samples. The fossilised birth-death process explicitly models the diversification, fossilisation, and sampling processes and naturally allows for sampled ancestors. This model was recently applied to estimate divergence times based on molecular data and fossil occurrence dates. We incorporate the fossilised birth-death model and a model of morphological trait evolution into a Bayesian total-evidence approach to dating species phylogenies. We apply this method to extant and fossil penguins and show that the modern penguins radiated much more recently than has been previously estimated, with the basal divergence in the crown clade occurring at~12.5 Ma and most splits leading to extant species occurring in the last 2 million years. Our results demonstrate that including stem-fossil diversity can greatly improve the estimates of the divergence times of crown taxa. The method is available in BEAST2 (v2.3) software www.beast2.org with packages SA (v1.1.3) and morph-models (v1.0.1) installed.
@article{gavryushkina2014,
author = {{Gavryushkina}, A. and {Heath}, T.~A. and {Ksepka}, D.~T. and {Stadler}, T. and {Welch}, D. and {Drummond}, A.~J.}, title = "{Bayesian total evidence dating reveals the recent crown radiation of penguins}", journal = {ArXiv e-prints}, archivePrefix = "arXiv", eprint = {1506.04797}, primaryClass = "q-bio.PE", keywords = {Quantitative Biology - Populations and Evolution}, year = 2015, month = jun, adsurl = {http://adsabs.harvard.edu/abs/2015arXiv150604797G} } |
|
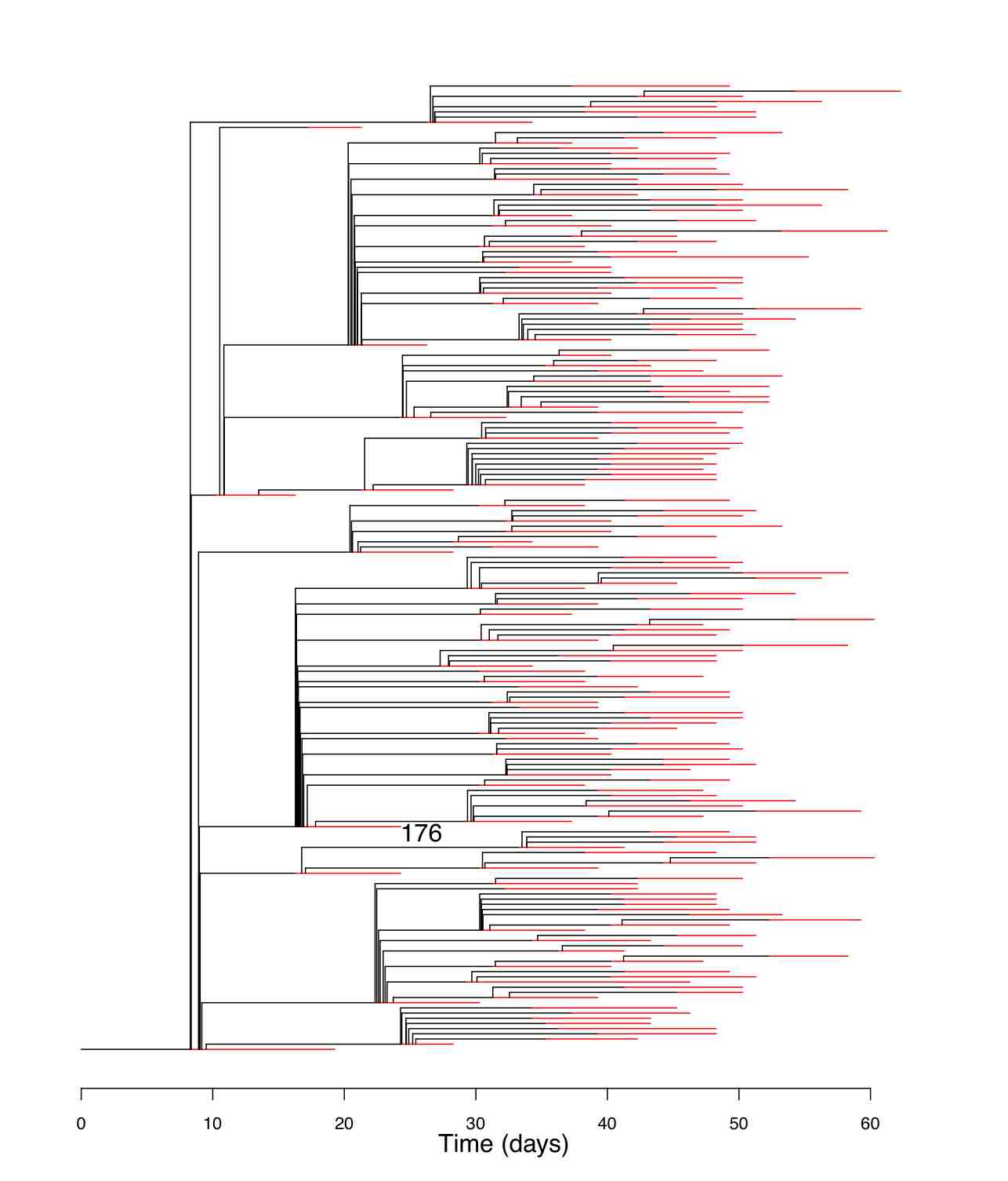
Bayesian Inference of Sampled Ancestor Trees for Epidemiology and Fossil Calibration
PLoS Computational Biology 10(12): e1003919, 2014
Phylogenetic analyses which include fossils or molecular sequences that are sampled through time require models that allow one sample to be a direct ancestor of another sample. As previously available phylogenetic inference tools assume that all samples are tips, they do not allow for this possibility. We have developed and implemented a Bayesian Markov Chain Monte Carlo (MCMC) algorithm to infer what we call sampled ancestor trees, that is, trees in which sampled individuals can be direct ancestors of other sampled individuals. We use a family of birth-death models where individuals may remain in the tree process after sampling, in particular we extend the birth-death skyline model [Stadler et al., 2013] to sampled ancestor trees. This method allows the detection of sampled ancestors as well as estimation of the probability that an individual will be removed from the process when it is sampled. We show that even if sampled ancestors are not of specific interest in an analysis, failing to account for them leads to significant bias in parameter estimates. We also show that sampled ancestor birth-death models where every sample comes from a different time point are non-identifiable and thus require one parameter to be known in order to infer other parameters. We apply our phylogenetic inference accounting for sampled ancestors to epidemiological data, where the possibility of sampled ancestors enables us to identify individuals that infected other individuals after being sampled and to infer fundamental epidemiological parameters. We also apply the method to infer divergence times and diversification rates when fossils are included along with extant species samples, so that fossilisation events are modelled as a part of the tree branching process. Such modelling has many advantages as argued in the literature. The sampler is available as an open-source BEAST2 package (https://github.com/CompEvol/sampled-ancestors)
@article{gavryushkina2014,
author = {Gavryushkina, Alexandra AND Welch, David AND Stadler, Tanja AND Drummond, Alexei J.}, journal = {PLoS Comput Biol}, publisher = {Public Library of Science}, title = {Bayesian Inference of Sampled Ancestor Trees for Epidemiology and Fossil Calibration}, year = {2014}, month = {12}, volume = {10}, url = {http://dx.doi.org/10.1371%2Fjournal.pcbi.1003919}, pages = {e1003919}, number = {12}, doi = {10.1371/journal.pcbi.1003919} } |
|
Modelling competition and dispersal in a statistical phylogeographic framework
Systematic Biology, 63(5): 743-752, 2014
Competition between organisms influences the processes governing the colonization of new habitats. As a consequence, species or populations arriving first at a suitable location may prevent secondary colonization. While adaptation to environmental variables (e.g., temperature, altitude, etc.) is essential, the presence or absence of certain species at a particular location often depends on whether or not competing species co-occur. For example, competition is thought to play an important role in structuring mammalian communities assembly. It can also explain spatial patterns of low genetic diversity following rapid colonization events or the “progression rule” displayed by phylogenies of species found on archipelagos. Despite the potential of competition to maintain populations in isolation, past quantitative analyses have largely ignored it because of the difficulty in designing adequate methods for assessing its impact. We present here a new model that integrates competition and dispersal into a Bayesian phylogeographic framework. Extensive simulations and analysis of real data show that our approach clearly outperforms the traditional Mantel test for detecting correlation between genetic and geographic distances. But most importantly, we demonstrate that competition can be detected with high sensitivity and specificity from the phylogenetic analysis of genetic variation in space.
@article{ranjard2014,
author = {Ranjard, Louis and Welch, David and Paturel, Marie and Guindon, Stéphane}, title = {Modelling competition and dispersal in a statistical phylogeographic framework}, volume = {63}, number = {5}, pages = {743-752}, year = {2014}, doi = {10.1093/sysbio/syu040}, URL = {http://sysbio.oxfordjournals.org/content/63/5/743.abstract}, eprint = {http://sysbio.oxfordjournals.org/content/63/5/743.full.pdf+html}, journal = {Systematic Biology} } |
|
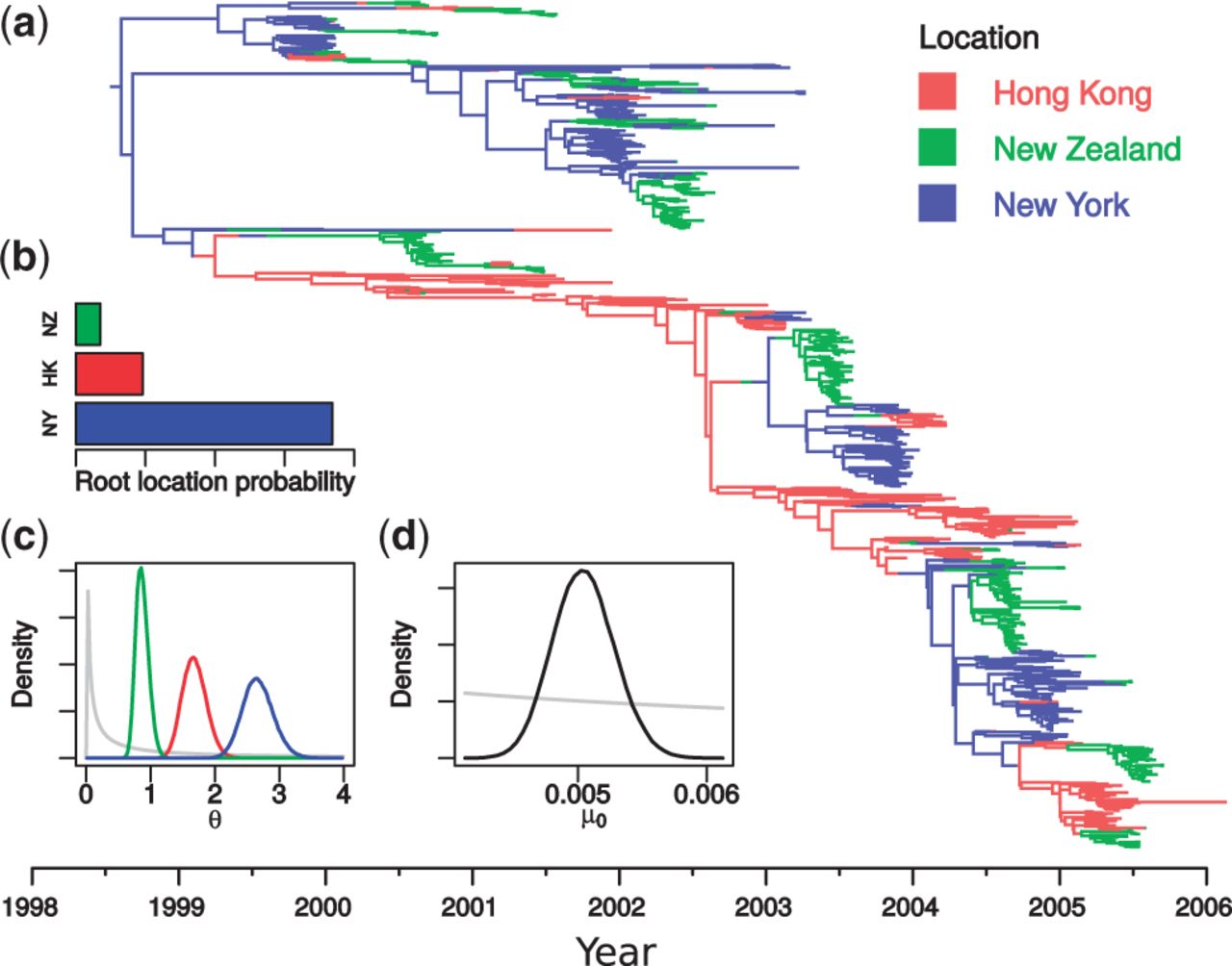
Efficient Bayesian inference under the structured coalescent
Bionformatics, 30(16): 2272-2279, 2014
Motivation: Population structure significantly affects evolutionary dynamics. Such structure may be due to spatial segregation, but may also reflect any other gene-flow-limiting aspect of a model. In combination with the structured coalescent, this fact can be used to inform phylogenetic tree reconstruction, as well as to infer parameters such as migration rates and subpopulation sizes from annotated sequence data. However, conducting Bayesian inference under the structured coalescent is impeded by the difficulty of constructing Markov Chain Monte Carlo (MCMC) sampling algorithms (samplers) capable of efficiently exploring the state space.
Results: In this article, we present a new MCMC sampler capable of sampling from posterior distributions over structured trees: timed phylogenetic trees in which lineages are associated with the distinct subpopulation in which they lie. The sampler includes a set of MCMC proposal functions that offer significant mixing improvements over a previously published method. Furthermore, its implementation as a BEAST 2 package ensures maximum flexibility with respect to model and prior specification. We demonstrate the usefulness of this new sampler by using it to infer migration rates and effective population sizes of H3N2 influenza between New Zealand, New York and Hong Kong from publicly available hemagglutinin (HA) gene sequences under the structured coalescent. Availability and implementation: The sampler has been implemented as a publicly available BEAST 2 package that is distributed under version 3 of the GNU General Public License at http://compevol.github.io/MultiTypeTree.
@Article{vaughan2014,
author = {Vaughan, Timothy G. and Kühnert, Denise and Popinga, Alex and Welch, David and Drummond, Alexei J.}, title = {Efficient Bayesian inference under the structured coalescent}, volume = {30}, number = {16}, pages = {2272-2279}, year = {2014}, doi = {10.1093/bioinformatics/btu201}, URL = {http://bioinformatics.oxfordjournals.org/content/30/16/2272.abstract}, eprint = {http://bioinformatics.oxfordjournals.org/content/30/16/2272.full.pdf+html}, journal = {Bioinformatics} } |
|
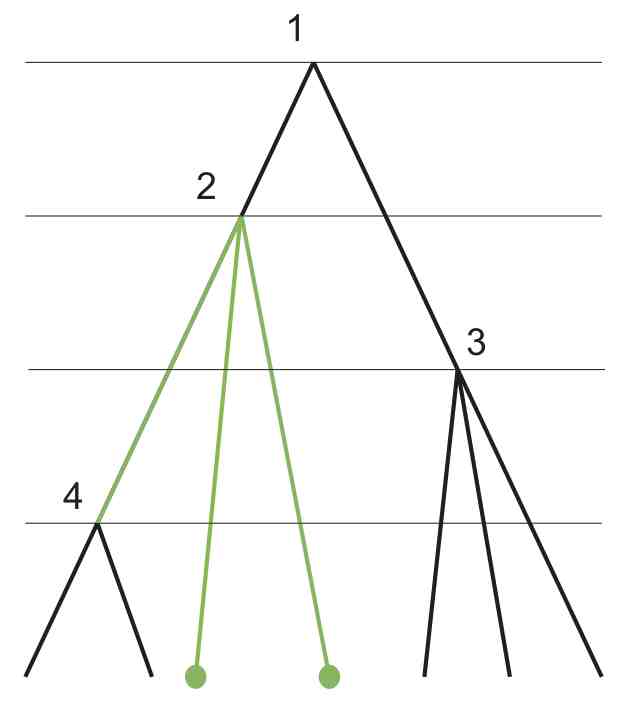
Recursive algorithms for phylogenetic tree counting
Algorithms for Molecular Biology 8:26, 2013
In Bayesian phylogenetic inference we are interested in distributions over a space of trees. The number of trees in a tree space is an important characteristic of the space and is useful for specifying prior distributions. When all samples come from the same time point and no prior information available on divergence times, the tree counting problem is easy. However, when fossil evidence is used in the inference to constrain the tree or data are sampled serially, new tree spaces arise and counting the number of trees is more difficult.
@Article{gavryushkina2013,
AUTHOR = {Gavryushkina, Alexandra and Welch, David and Drummond, Alexei}, TITLE = {Recursive algorithms for phylogenetic tree counting}, JOURNAL = {Algorithms for Molecular Biology}, VOLUME = {8}, YEAR = {2013}, NUMBER = {1}, PAGES = {26}, URL = {http://www.almob.org/content/8/1/26}, DOI = {10.1186/1748-7188-8-26}, PubMedID = {24164709} } |
|
A Network-based Analysis of the 1861 Hagelloch Measles Data
Biometrics, 68 (3):755-765, 2012
In this article, we demonstrate a statistical method for fitting the parameters of a sophisticated network and epidemic model to disease data.
The pattern of contacts between hosts is described by a class of dyadic independence exponential-family random graph models (ERGMs), whereas
the transmission process that runs over the network is modeled as a stochastic susceptible-exposed-infectious-removed (SEIR) epidemic.
We fit these models to very detailed data from the 1861 measles outbreak in Hagelloch, Germany.
The network models include parameters for all recorded host covariates including age, sex, household, and classroom membership and household
location whereas the SEIR epidemic model has exponentially distributed transmission times with gamma-distributed latent and infective periods.
This approach allows us to make meaningful statements about the structure of the population—separate from the transmission process—as well as
to provide estimates of various biological quantities of interest, such as the effective reproductive number, R. Using reversible jump Markov chain Monte Carlo,
we produce samples from the joint posterior distribution of all the parameters of this model—the network, transmission tree, network parameters,
and SEIR parameters—and perform Bayesian model selection to find the best-fitting network model. We compare our results with those of previous
analyses and show that the ERGM network model better fits the data than a Bernoulli network model previously used. We also provide a software package,
written in R, that performs this type of analysis.
@article{groendyke2012network,
title={A Network-based Analysis of the 1861 Hagelloch Measles Data}, author={Groendyke, Chris and Welch, David and Hunter, David R}, journal={Biometrics}, volume={68}, number={3}, pages={755--765}, year={2012}, doi={10.1111/j.1541-0420.2012.01748.x}, publisher={Wiley Online Library} } |
|
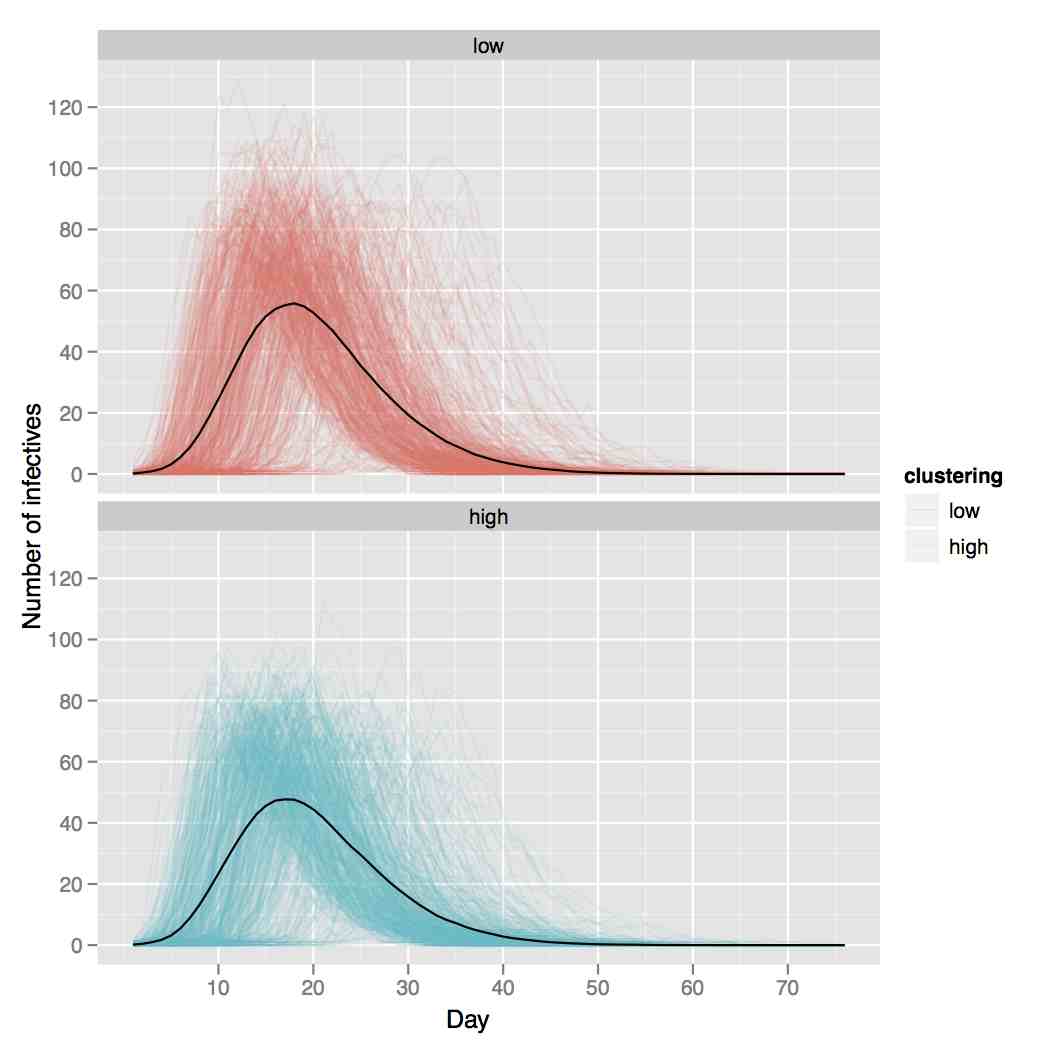
Is network clustering detectable in transmission trees?
Viruses 3 (6), 659-676, 2011
Networks are often used to model the contact processes that allow pathogens to spread between hosts but it remains unclear which models best describe these networks. One question is whether clustering in networks, roughly defined as the propensity for triangles to form, affects the dynamics of disease spread. We perform a simulation study to see if there is a signal in epidemic transmission trees of clustering. We simulate susceptible-exposed-infectious-removed (SEIR) epidemics (with no re-infection) over networks with fixed degree sequences but different levels of clustering and compare trees from networks with the same degree sequence and different clustering levels. We find that the variation of such trees simulated on networks with different levels of clustering is barely greater than those simulated on networks with the same level of clustering, suggesting that clustering can not be detected in transmission data when re-infection does not occur.
@article{welch2011,
Author = {Welch, David}, Doi = {10.3390/v3060659}, Issn = {1999-4915}, Journal = {Viruses}, Number = {6}, Pages = {659--676}, Pubmedid = {21731813}, Title = {Is Network Clustering Detectable in Transmission Trees?}, Url = {http://www.mdpi.com/1999-4915/3/6/659}, Volume = {3}, Year = {2011} } |
|
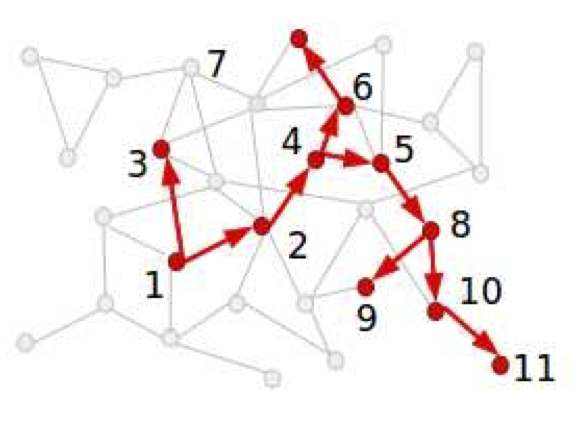
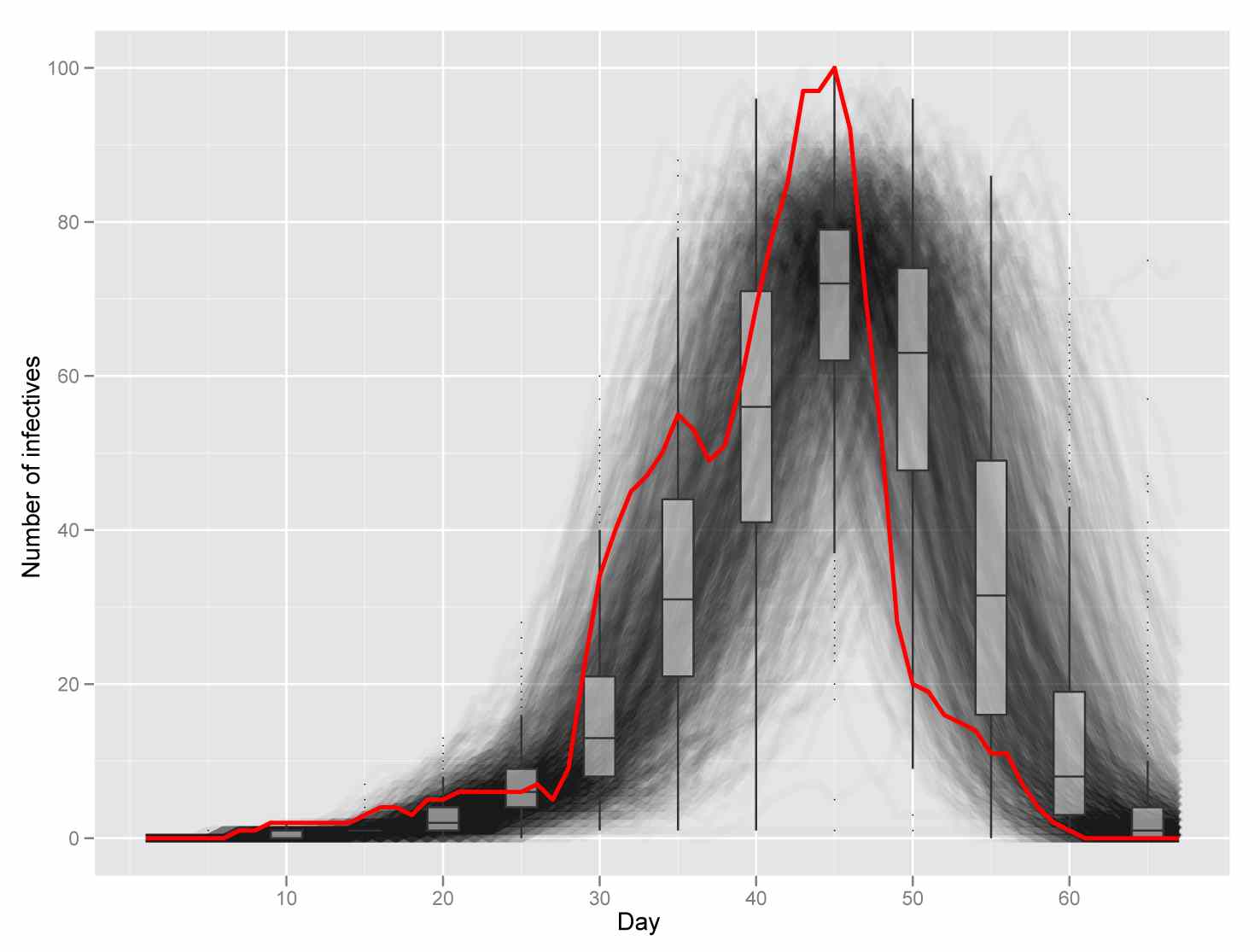
Statistical inference to advance network models in epidemiology
Epidemics, 3(1), 38-45, 2011
Contact networks are playing an increasingly important role in the study of epidemiology. Most of the existing work in this area has focused on considering the effect of underlying network structure on epidemic dynamics by using tools from probability theory and computer simulation. This work has provided much insight on the role that heterogeneity in host contact patterns plays on infectious disease dynamics. Despite the important understanding afforded by the probability and simulation paradigm, this approach does not directly address important questions about the structure of contact networks such as what is the best network model for a particular mode of disease transmission, how parameter values of a given model should be estimated, or how precisely the data allow us to estimate these parameter values. We argue that these questions are best answered within a statistical framework and discuss the role of statistical inference in estimating contact networks from epidemiological data.
@article{Welch201138,
title = "Statistical inference to advance network models in epidemiology ", journal = "Epidemics ", volume = "3", number = "1", pages = "38 - 45", year = "2011", note = "", issn = "1755-4365", doi = "http://dx.doi.org/10.1016/j.epidem.2011.01.002", url = "http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3090691/pdf/nihms-276851.pdf", author = "David Welch and Shweta Bansal and David R. Hunter", keywords = "Contact networks", keywords = "Statistical inference", keywords = "Transmission networks", keywords = "Estimation " } |
|
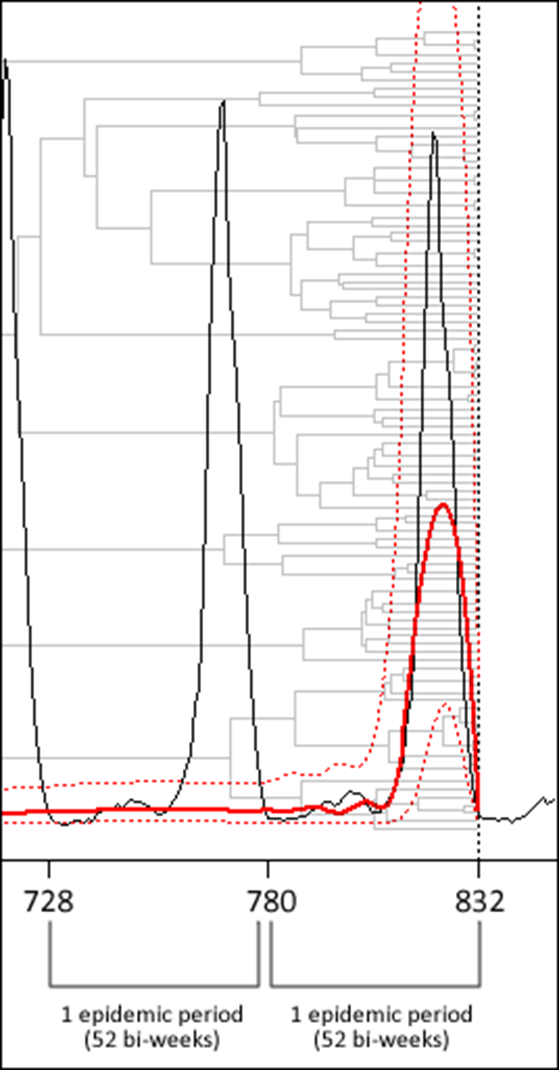
Bayesian inference for contact networks given epidemic data
Scandinavian Journal of Statistics 38 (3), 600-616, 2011
In this article, we estimate the parameters of a simple random network and a stochastic epidemic on that network using data consisting of recovery times of infected hosts. The SEIR epidemic model we fit has exponentially distributed transmission times with Gamma distributed exposed and infectious periods on a network where every edge exists with the same probability, independent of other edges. We employ a Bayesian framework and Markov chain Monte Carlo (MCMC) integration to make estimates of the joint posterior distribution of the model parameters. We discuss the accuracy of the parameter estimates under various prior assumptions and show that it is possible in many scientifically interesting cases to accurately recover the parameters. We demonstrate our approach by studying a measles outbreak in Hagelloch, Germany, in 1861 consisting of 188 affected individuals. We provide an R package to carry out these analyses, which is available publicly on the Comprehensive R Archive Network.
@article {groendyke2011,
author = {GROENDYKE, CHRIS and WELCH, DAVID and HUNTER, DAVID R.}, title = {Bayesian Inference for Contact Networks Given Epidemic Data}, journal = {Scandinavian Journal of Statistics}, volume = {38}, number = {3}, publisher = {Blackwell Publishing Ltd}, issn = {1467-9469}, url = {http://dx.doi.org/10.1111/j.1467-9469.2010.00721.x}, doi = {10.1111/j.1467-9469.2010.00721.x}, pages = {600--616}, keywords = {Erdős-Rényi, exponential random graph model (ERGM), MCMC, measles, stochastic SEIR epidemic}, year = {2011}, } |
|
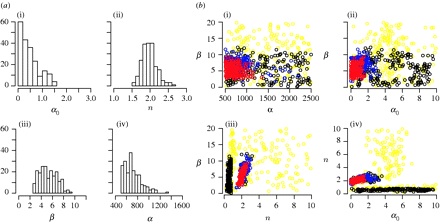
Protocols for sampling viral sequences to study epidemic dynamics
Journal of the Royal Society Interface, 7, 1119-1127, 2010
With more emphasis being put on global infectious disease monitoring, viral genetic data are being collected at an astounding rate, both within and without the context of a long-term disease surveillance plan. Concurrent with this increase have come improvements to the sophisticated and generalized statistical techniques used for extracting population-level information from genetic sequence data. However, little research has been done on how the collection of these viral sequence data can or does affect the efficacy of the phylogenetic algorithms used to analyse and interpret them. In this study, we use epidemic simulations to consider how the collection of viral sequence data clarifies or distorts the picture, provided by the phylogenetic algorithms, of the underlying population dynamics of the simulated viral infection over many epidemic cycles. We find that sampling protocols purposefully designed to capture sequences at specific points in the epidemic cycle, such as is done for seasonal influenza surveillance, lead to a significantly better view of the underlying population dynamics than do less-focused collection protocols. Our results suggest that the temporal distribution of samples can have a significant effect on what can be inferred from genetic data, and thus highlight the importance of considering this distribution when designing or evaluating protocols and analysing the data collected thereunder.
@article{Stack2010,
author = {Stack, J. Conrad and Welch, J. David and Ferrari, Matt J. and Shapiro, Beth U. and Grenfell, Bryan T.}, title = {Protocols for sampling viral sequences to study epidemic dynamics}, year = {2010}, doi = {10.1098/rsif.2009.0530}, URL = {http://rsif.royalsocietypublishing.org/content/early/2010/02/08/rsif.2009.0530.abstract}, eprint = {http://rsif.royalsocietypublishing.org/content/early/2010/02/08/rsif.2009.0530.full.pdf+html}, journal = {Journal of The Royal Society Interface} } |
|
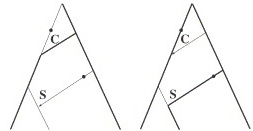
Approximate Bayesian Computation scheme for parameter inference and model selection in dynamical systems
Journal of the Royal Society Interface 6, 187-202, 2009
Approximate Bayesian computation (ABC) methods can be used to evaluate posterior distributions without having to calculate likelihoods. In this paper, we discuss and apply an ABC method based on sequential Monte Carlo (SMC) to estimate parameters of dynamical models. We show that ABC SMC provides information about the inferability of parameters and model sensitivity to changes in parameters, and tends to perform better than other ABC approaches. The algorithm is applied to several well-known biological systems, for which parameters and their credible intervals are inferred. Moreover, we develop ABC SMC as a tool for model selection; given a range of different mathematical descriptions, ABC SMC is able to choose the best model using the standard Bayesian model selection apparatus.
@article{Toni2009,
author = {Toni, Tina and Welch, David and Strelkowa, Natalja and Ipsen, Andreas and Stumpf, Michael P.H}, title = {Approximate Bayesian computation scheme for parameter inference and model selection in dynamical systems}, volume = {6}, number = {31}, pages = {187-202}, year = {2009}, doi = {10.1098/rsif.2008.0172}, URL = {http://rsif.royalsocietypublishing.org/content/6/31/187.abstract}, eprint = {http://rsif.royalsocietypublishing.org/content/6/31/187.full.pdf+html}, journal = {Journal of The Royal Society Interface} } |
|
Integrating genealogy and epidemiology: The ancestral infection and selection graph as a model for reconstructing host virus histories.
Theoretical Population Biology. 68: 65-75, 2005
We model the genealogies of coupled haploid host–virus populations. Hosts reproduce and replace other hosts as in the Moran model. The virus can be transmitted between individuals of the same and succeeding generations. The epidemic model allows a selective advantage for susceptible over infected hosts. The coupled host–virus ancestry of a sample of hosts is embedded in a branching and coalescing structure that we call the Ancestral Infection and Selection Graph, a direct analogue to the Ancestral Selection Graph of Krone and Neuhauser [1997. Theoret. Population Biol. 51, 210–237]. We prove this and discuss various special cases. We show that the inter-host viral genealogy is a scaled coalescent. Using simulations, we compare the viral genealogy under this model to earlier published models and investigate the estimatability of the selection and infectious contact rates. We use simulations to compare the persistence of the disease with the time to the ultimate ancestor.
@article{Welch2005,
title = "Integrating genealogy and epidemiology: The ancestral infection and selection graph as a model for reconstructing host virus histories ", journal = "Theoretical Population Biology ", volume = "68", number = "1", pages = "65 - 75", year = "2005", note = "John Maynard Smith Memorial Issue ", issn = "0040-5809", doi = "http://dx.doi.org/10.1016/j.tpb.2005.03.003", url = "http://www.sciencedirect.com/science/article/pii/S0040580905000511", author = "David Welch and Geoff K. Nicholls and Allen Rodrigo and Wiremu Solomon" } |
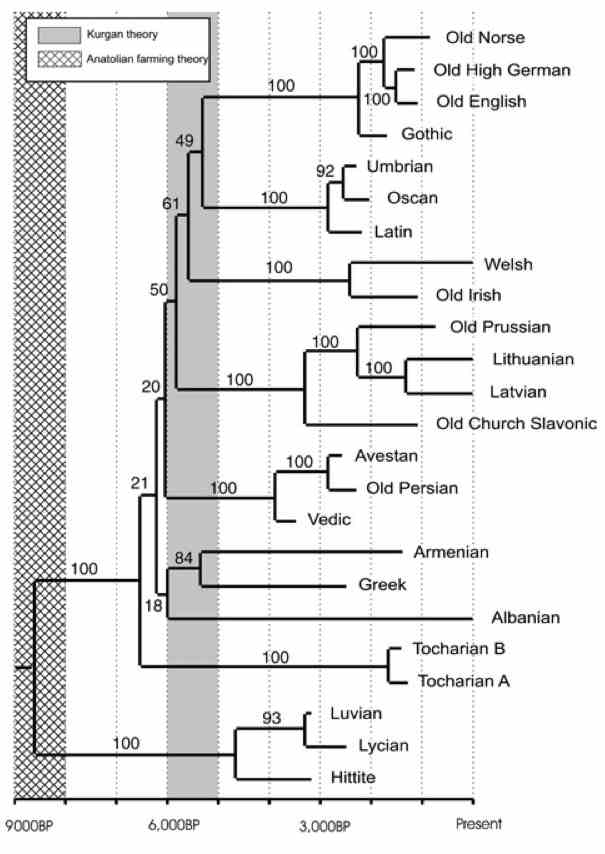
|
From Words to Dates: Water Into Wine, Mathemagic or Phylogenetic Inference?
Quantitative Methods in Language Comparison. Special issue of Transactions of the Philological Society, McMahon, A. (Ed) 103.2, 2005
Gray & Atkinson's (2003) application of quantitative phylogenetic methods to Dyen, Kruskal & Black's (1992) Indo-European database produced controversial divergence time estimates. Here we test the robustness of these results using an alternative data set of ancient Indo-European languages. We employ two very different stochastic models of lexical evolution – Gray & Atkinson's (2003) finite-sites model and a stochastic-Dollo model of word evolution introduced by Nicholls & Gray (in press). Results of this analysis support the findings of Gray & Atkinson (2003). We also tested the ability of both methods to reconstruct phylogeny and divergence times accurately from synthetic data. The methods performed well under a range of scenarios, including widespread and localized borrowing.
@article {atkinson2005,
author = {Atkinson, Quentin and Nicholls, Geoff and Welch, David and Gray, Russell}, title = {From words to dates: water into wine, mathemagic or phylogenetic inference?}, journal = {Transactions of the Philological Society}, volume = {103}, number = {2}, publisher = {Blackwell Publishing}, issn = {1467-968X}, url = {http://dx.doi.org/10.1111/j.1467-968X.2005.00151.x}, doi = {10.1111/j.1467-968X.2005.00151.x}, pages = {193--219}, year = {2005} } |
Software
|
EpiNet: An R package for fitting network models to epidemic dataWritten by Chris Groendyke, David Welch and David Hunter (2011). An R package that simulates transmission of diseases through contact networks and performs Bayesian inference on network and epidemic parameters for given epidemic data. |
|
TraitLab: Software for simulating and fitting tree-like binary trait data in MatlabWritten by Geoff Nicholls, David Welch, Robin Ryder (2010). An easy-to-use package for simulation, analysis and phylogenetic inference on binary (absence-presence) trait data. |